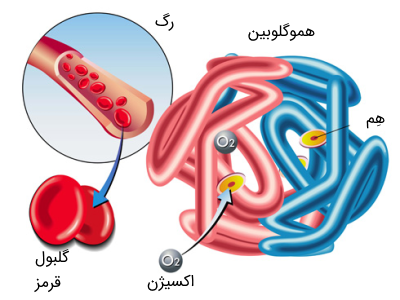
در ادامه تشریح تالاسمی چیست؟ همانطور که میدانید، هموگلوبین که از دوزنجیره پروتئینی آلفا و بتا تشکیل شده است، به عنوان جزء حامل اکسیژن در گلبول قرمز عمل میکند. اگر یکی از این دو پروتئین در بدن به اندازه کافی تولید نشود، گلبولهای قرمز به درستی تشکیل نشده و درنتیجه نمیتواند اکسیژن را به میزان کافی در خون حمل کند.
این اتفاق باعث ایجاد نوعی کم خونی میشود که از اوایل کودکی شروع شده و در طول زندگی فرد ادامه مییابد. لازم به ذکر است که تالاسمی نوعی بیماری ارثی است، به این معنا که باید حداقل یکی از والدین ناقل این بیماری باشد. این بیماری به دلیل یک جهش ژنتیکی یا حذف برخی از قطعات ژنی کلیدی ایجاد می شود.
به دلیل حذف ژن آلفا گلوبین ایجاد میشود که منجر به کاهش یا عدم تولید زنجیره آلفا گلوبین در هوگلوبین میشود. ژن آلفا گلوبین دارای 4 آلل (جزء) است و شدت بیماری بسته به تعداد حذف آللها از خفیف تا شدید متغیر است.
حذف چهار آلل شدیدترین شکل بیماری است و در آن بطور کلی آلفا گلوبین تولید نمیشود و زنجیره های پروتئینی گامای اضافی (که در دوران جنینی در هموگلوبین وجود دارند) ساختارهایی در هموگلوبین تشکیل میدهند که منجر به هیدروپس جنینی و درنتیجه مغایرت با حیات جنین می شود.
تالاسمی بتا ناشی از جهش نقطه ای در ژن بتا گلوبین است و بر اساس انواع جهشهای ژن بتا، به سه دسته تقسیم میشود:
1. یک جهش هتروزیگوت (یک ژن جهش یافته) منجر به بتا تالاسمی مینور میشود که در آن زنجیرههای بتا به میزان کافی تولید نمیشوند. این حالت خفیف و معمولاً بدون علامت است.
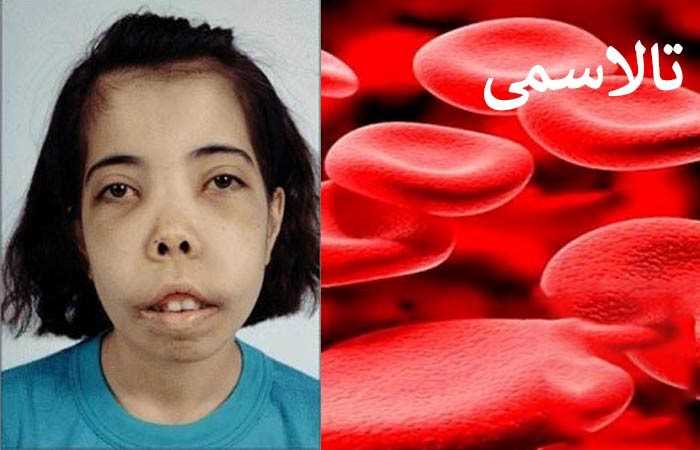
2. بتا تالاسمی ماژور یا ک خونی کولی، در اثر یک جهش هموزیگوت (دو ژن جهش یافته) در ژن بتا گلوبین ایجاد میشود که در نتیجه باعث غیاب کامل (عدم تشکیل) زنجیرههای بتا میشود. این حالت از نظر بالینی علائمی دارد مثل زردی، تاخیر در رشد، هپاتواسپلنومگالی (بزرگی طحال و کبد)، ناهنجاریهای غدد و هورمونها و کم خونی شدید که نیاز به تزریق مادام العمر خون دارد.
3. وضعیت بین دو حالت فوق، نوع بتا تالاسمی اینترمدیا (بینابینی) نامیده میشود که علائم بالینی خفیف تا متوسط دارند. در این حالت زنجیرههای آلفا گلوبین اضافه تجمع یافته و سپس رسوب میکنند و به غشای گلبولهای قرمز آسیب رسانده، منجر به همولیز (تجزیه خون) در داخل عروق میشوند. این مرگ زودرس سلول های مسئول خون سازی منجر به خون سازی غیر موثر میشود.
مکانیسم بیماری تالاسمی(Thalassemia):
هموگلوبین یک تترامر (ساخار چهار جزئی) متشکل از دو زنجیره آلفا گلوبین و دو زنجیره غیر آلفا گلوبین است. هموگلوبین انواعی دارد از جمله: هموگلوبین جنینی (HbF) که هموگلوبین اولیه تا شش ماهگی است و از دو زنجیره آلفا و دو زنجیره گاما تشکیل شده است. هموگلوبین بزرگسالان که در درجه اول هموگلوبین (HbA)A است و از دو زنجیره آلفا و دو زنجیره بتا تشکیل شده است.
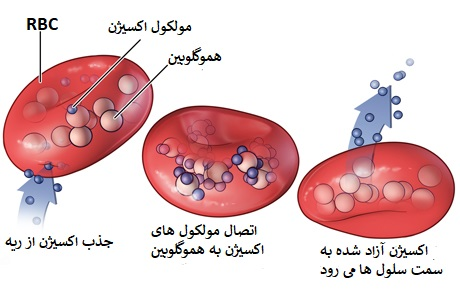
در بتا تالاسمی دو اتفاق میافتد: اول، کاهش سنتز هموگلوبین که باعث کم خونی و همچنین افزایش HbF و HbA2 میشود زیرا میزان زنجیره های بتای مورد نیاز برای تشکیل HbA ، کاهش مییابد. دوم، زنجیرههای آلفای اضافی نسبی، ساختارهای نامحلولی را تشکیل میدهند که باعث همولیز (تجزیه خون) میشوند.
این خون ریزی غیرموثر منجر به کم خونی شدید و هیپرپلازی اریتروئید (افزایش تعداد سلولهای خون ساز در مغز استخوان) همراه با انبساط مغز استخوان و خون سازی خارج از مغز استخوان میشود.
انبساط مغز استخوان باعث ناهنجاریهای استخوانی می شود که مشخصهی آن، تغییر شکل استخوانهای صورت، برجستگی پیشانی و بیرونزدگی فک بالا است.
همچنین سیگنال دهی ناشی از انبساط مغز استخوان تولید هپسیدین را مهار کرده و باعث جذب بیش از حد آهن میشود (در مورد هپسیدین و عملکردآن، در مقالهی کم خونی توضیح مفصلی داده شده است). بیمارانی که به اندازه کافی درمان نشده اند و بیماران وابسته به تزریق خون در معرض خطر اضافه بار آهن به اندامها هستند.
بتا تالاسمی مینور باعث کم خونی خفیف در نتیجهی کاهش سنتز هموگلوبین A میشود. افراد مبتلا به بتا تالاسمی مینور دارای یک ژن فاقد عملکرد درست هستند، بنابراین آنها هنوز هم میتوانند هموگلوبین کافی برای تأمین نیاز منظم بدن بدون ایجاد مشکل قابل توجه در سلولهای تولید کننده گلبول قرمز را تولید کنند.
علاوه بر این، کمبود هموگلوبین A با افزایش سایر اشکال هموگلوبین، مثلا HbA2 جبران می شود.
علت ایجاد بیماری تالاسمی چیست؟

همانطور که پیش تر ذکر شد، تالاسمی نوعی بیماری اتوزومال مغلوب است، یعنی برای انتقال بیماری به فرزندان هر دو والدین باید مبتلا یا ناقل بیماری باشند. این بیماری در اثر جهش یا حذف ژنهای هموگلوبین ایجاد میشود که درنهایت منجر به تولید کم یا عدم وجود زنجیرههای آلفا یا بتا میشود. بیش از 200 جهش به عنوان دلایل احتمالی ایجاد تالاسمی شناسایی شده اند. تالاسمی آلفا در اثر حذف ژنهای آلفا گلوبین و تالاسمی بتا در اثر وقوع جهش نقطهای در ژن بتا گلوبین در کروموزوم 11 ایجاد میشوند.
جالب است بدانید که تالاسمی آلفا در کشورهای آسیایی و آفریقایی شایع است در حالیکه بتا تالاسمی در جمعیت مدیترانهای شایع تر است.
علائم بیماری تالاسمی:
تظاهرات تالاسمی بسته به نوع و شدت آن بسیار متفاوت است. بطور کلی نشانه های احتمالی تالاسمی عبارتند از:
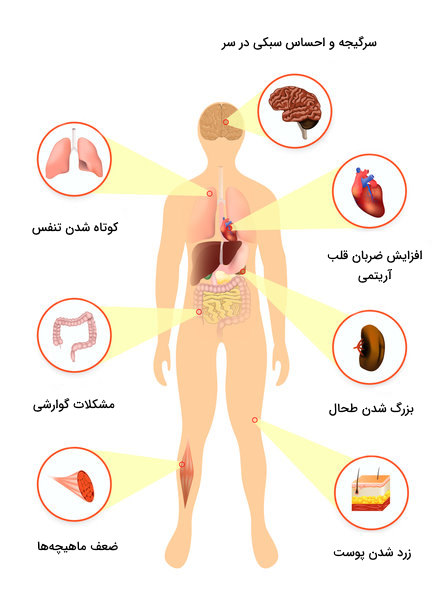
• رنگ پریدگی پوست به دلیل کمخونی و یا زردی آن به دلیل افزایش بیلی روبینمی در خون به دنبال افزایش تجزیه خون داخل عروق – زخم روی اندامها – برنزه شدن پوست بهدلیل رسوب مزمن آهن
• بیماران ممکن است خستگی ناشی از کم خونی را به عنوان اولین شکایت گزارش کنند
• گسترش خون سازی در مناطق خارج از مفز استخوانهای اصلی، منجر به تغییر شکل استخوانهای صورت و سایر استخوانها و صورتی به نام صورت سنجاب میشود.
• رسوب آهن در سلولهای ماهیچه ای قلبی در اثر تزریق مداوم و طولانی مدت خون، میتواند ریتم طبیعی قلب را مختل کند. همچنین به دلیل کم خونی مزمن، نارسایی قلبی نیز ممکن است ایجاد شود.
• افزایش مزمن بیلی روبین خون می تواند منجر به ایجاد سنگ صفراوی از جنس بیلی روبین شود که به صورت درد کولیکی (ماهیت درد تقریبا مشابه درد ناشی از سنگ کلیه است) ظاهر میشود. همچنین رسوب مزمن آهن و گسترش خون سازی (خارج از مناطق اصلیِ مغز استخوان) در کبد و طحال، ممکن است باعث افزایش سایز آنها شود. بعلاوه، ممکن است مزمن شدن همولیز (تجزیه خون) باعث انفارکتوس طحال و از بین رفتن خود به خودی آن نیز بشود.
• درگیری کبد به دلیل نیاز مزمن به تزریق خون یک یافته رایج در تالاسمی است. نارسایی مزمن کبدی یا سیروز (چروک شدن کبد) ممکن است بعلت رسوب آهن یا هپاتیت ویروسی (مرتبط با انتقال خون) رخ دهد.
• کم خونی باعث کاهش سرعت رشد کودک میشود، بنابراین تالاسمی میتواند باعث تاخیر در بلوغ کودک شود.
• اضافه بار آهن میتواند منجر به رسوب آن در دستگاههای مختلف بدن و در نهایت اختلال عملکرد آنها شود. رسوب آهن در پانکراس منجر به دیابت، رسوب آن در غدد تیروئید یا پاراتیروئید به ترتیب منجر به کم کاری تیروئید و کم کاری پاراتیروئید، رسوب آن در مفاصل منجر به التهاب و دردهای مفصلی مزمن، رسوب آن در مغز، منجر به ایجاد بیماری پارکینسون بصورت زودرس میشود.
• بتا تالاسمی مینور معمولاً به طور اتفاقی در آزمایش خون کشف می شود. بیماران اغلب علائم خفیف کم خونی، بدون یافته های معاینه فیزیکی قابل توجه دارند.
• بیماران مبتلا به بتا تالاسمی ماژور، اگر تشخیص قبل از تولد و طی غربالگریهای ژنتیکی و دوران بارداری مشخص نشده باشد، بین 6 تا 24 ماهگی زمانی که تولید هموگلوبین از جنین (HbF) به بزرگسال (HbA) تغییر میکند، به دلیل کم خونی شدید مراجعه میکنند. این نوزادان اغلب مشکلات تغذیه، تحریک پذیری، عدم رشد، رنگ پریدگی، اسهال، حملات مکرر تب و بزرگ شدن شکم ناشی از بزرگی کبد و طحال دارند.
نوزادانی که تحت درمان قرار نگرفته اند، از کندی شدید رشد، زردی، لکههای قهوهای روی پوست، عضلات اسکلتی ضعیف، ژنو والگوم (زانوها بصورت حرف X در می آیند)، زخمهای ساق پا و تغییر شکل استخوانی رنج میبرند.
درمان تالاسمی:
درمان تالاسمی به نوع و شدت بیماری بستگی دارد:
- در تالاسمی خفیف (که هموگلوبین بین 6 تا 10 گرم بر دسی لیتر است)، علائم و نشانهها خفیف بوده و درمان زیادی نیاز ندارند. گاهی اوقات، در شرایطی مانند زایمان یا بعد از هرنوع جراحی، ممکن است بیمار نیاز به تزریق خون داشته باشد.
- تالاسمی متوسط تا شدید (هموگلوبین بین 5 تا 6 گرم در دسی لیتر):
1. انتقال خون مکرر: اشکال شدیدتر تالاسمی اغلب نیاز به تزریق خون منظم، احتمالاً هر چند هفته یکبار دارد.
2. به دلیل تزریق طولانی مدت خون، آهن افزایش یافته و شروع به رسوب در اندامهای مختلف بدن میکند. به همین دلیل داروهای چلاتور آهن (مانند دفراسیروکس، دفروکسامین، دفریپرون) به طور همزمان برای حذف آهن اضافی از بدن استفاده میشوند.
3. پیوند سلولهای بنیادی: پیوند سلولهای بنیادی، (پیوند مغز استخوان)، درمواردی مانند کودکانی که با تالاسمی شدید متولد میشوند، ممکن است نیاز به انتقال خون مادام العمر را در کودک برطرف کند. با این حال، این روش عوارض و خطرات خاص خود را دارد.
4. ژن درمانی: این درمان، آخرین پیشرفت در مدیریت بیماری تالاسمی شدید است. ژن درمانی شامل برداشتن سلولهای بنیادی خونساز از بیمار و اصلاح ژنتیکی آنها، و سپس تزریق مجدد سلولهای اصلاح شده به بدن بیمار است.
5. اسپلنکتومی: بیماران مبتلا به تالاسمی ماژور اغلب تحت عمل جراحی برداشت طحال قرار میگیرند تا تعداد تزریقات خون کم شود. اسپلنکتومی نه تنها تعداد تزریقهای مورد نیاز را محدود میکند، بلکه گسترش خون سازی خارج از مغز استخوان را نیز کنترل میکند. ایمن سازی و واکسیناسیون بعد از عمل جراحی برداشتن طحال، برای جلوگیری از عفونتهای باکتریال ضروری است. لازم به ذکر است احتمال ابتلا به عفونت گسترده بدن بعد از برداشتن طحال در کودکان وجود دارد، بنابراین این عمل جراحی تا 6 الی 7 سالگی به تعویق میافتد.
6. کوله سیستکتومی: در بیماران مبتلا به تالاسمی، به دلیل افزایش تجزیه هموگلوبین و رسوب بیلی روبین در کیسه صفرا، ممکن است به سنگ صفراوی مبتلا شوند. در صورت علامت دار شدن، بیماران باید همزمان با انجام اسپلنکتومی، تحت عمل جراحی برداشتن کیسه صفرا نیز قرار گیرد/
عوارض بیماری تالاسمی:
تالاسمی می تواند عوارض زیر را ایجاد کند:
• یرقان (زردی) و سنگهای صفراوی ناشی از افزایش بیلی روبین خون
• نازک شدن قشر مغز و اعوجاج استخوانها
• نارسایی بالای قلبی بهدلیل کم خونی شدید و آریتمی (اختلال ریتم قلبی) – لازم به ذکر است که درگیری قلبی علت اصلی مرگ و میر در بیماران مبتلا به تالاسمی است.
• هپاتواسپلنومگالی (بزرگی کبد و طحال) ناشی از خونسازی خارج مدولاری (خارج از مغز استخوانهای اصلی مسئول خونسازی) و رسوب آهن اضافی به دلیل تزریق مداوم خون
• رسوب آهن اضافی میتواند منجر به مشکلاتی مانند اختلالات غدد و هورمونها، مشکلات مفصلی، تغییر رنگ پوست شود.
• عوارض عصبی مانند نوروپاتی محیطی
• کاهش سرعت رشد و تاخیر در بلوغ
• حوادث ترومبوآمبولیک (لخته شدن خون) مانند ترومبوز وریدی عمقی (لخته شدن خون در وریدهای پا)، آمبولی ریوی و انسداد مکرر شریانی در بیماران تالاسمی بیشتر از جمعیت عادی است.
• سایر عوارض عبارتند از هپاتیت مزمن (ناشی از عفونت با ویروس های هپاتیت B و C بهدلیل انتقال خون)، سیروز (بهدلیل رسوب بیش از حد آهن در کبد)، عفونت HIV (ایدز) و پوکی استخوان.
پیشگیری از ایجاد عوارض بیشتر:
1) بهتر است برنامه ی درمانی مناسب تحت نظر پزشک و رژیم غذایی و عادتهای زندگی سالم، اتخاذ کنید.
2) از مصرف خودسرانهی آهن اضافی (بطور مثال مولتی ویتامین ها یامکمل های خوراکی حاوی آهن) خودداری کنید.
3) رژیم غذایی سالم داشته باشید. مصرف رژیم غذایی متعادل و سالم حاوی مقدار زیادی غذاهای مغذی، می تواند به شما در داشتن احساس بهتر، کمک کند و انرژی را افزایش دهد. گاهی اوقات پزشک معالج مصرف مکمل اسید فولیک را برای کمک به ساخت گلبول های قرمز جدید توصیه میکنند
4) سعی کنید از بدن خود در برابر ابتلا به انواع عفونتها محافظت کنید. واکسن سالانه آنفولانزا، مننژیت، پنوموکوک و هپاتیت B برای جلوگیری از عفونت توصیه میشود.
سخن پایانی:
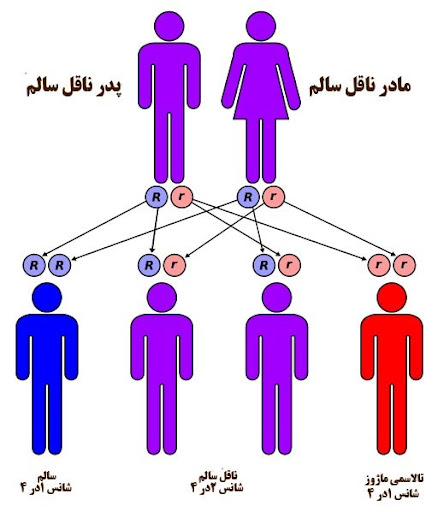
خوب است بدانید بیماری تالاسمی، ماهیت ارثی دارد به گونهای که اگر پدر و مادر هر دو مبتلا به تالاسمی مینور باشند، یک چهارم ممکن است فرزند آنها مبتلا به تالاسمی ماژور شود. امروزه غربالگریهای ژنتیکی قبل از ازدواج و قبل از بارداری و همچنین در ماههای اول بارداری، برای تشخیص بیماریهای ارثی مهم مانند تالاسمی ماژور بسیار کمک کننده و مفید هستند.
بنابراین برای جلوگیری از ایجاد بیماریای ارثی صعب العلاج در فرزندان، بهتر است آزمایشات ژنتیکی مربوطه انجام شود. چرا که بیماریهای ارثی مانند تالاسمی، نه تنها درمان قطعی خاصی ندارند و بعلاوه، نیاز به پیگیری مداوم داشته و هزینههای مالی و روانی زیادی بر خانواده و جامعه تحمیل میکنند.
سلامتی همیشگی شما، آرزوی ماست : تیم لاویا